

Apresentamos, no quadro abaixo, os itens que destacaremos na apresentação de Hemostasia. Se você desejar ir a algum assunto é só clicar no item correspondente.
INTRODUÇÃO
Nessa página, denominada Hemostasia 1, vamos considerar a hemostasia obedecendo à ordem histórica de seu desenvolvimento. Por esta razão, iniciaremos o assunto expondo o modelo denominado "MODELO DA COAGULAÇÃO EM CASCATA".
Na página, Hemostasia 2, mostraremos o modelo hemostático sob a visão do "MODELO CELULAR DA HEMOSTASIA", com ênfase às reações acontecendo nas membranas de diversas células.
MODELO DA COAGULAÇÃO EM CASCATA
O primeiro modelo para a coagulação foi descrito por vários autores, com destaque para Margolis, Ratnoff e Macfarlane:
[1] MargolisJ. Initiation Of Blood Coagulation By Glass And Related Surfaces. J. Physiol. I957; I37, 95-IO9.
[2] Macfarlane RG. An enzyme cascade in the blood clotting mechanism, and its function as a biological amplifier. Nature1964;202:498–9.
[3] Davie EW, Ratnoff OD. Waterfall sequence for intrinsic blood clotting. Science 1964;145: 1310–2.
A presente explicação, derivada de uma comunicação anterior (Margolis, 1956), sugere que as alterações observadas na coagulação do sangue, são o resultado da exposição a superfícies tais como vidro, caulim e várias formas de sílica, que são conhecidos por ativar a coagulação.
Hoje, sabemos que, in vivo, a ativação do FXII se dá em contato com os tecidos expostos devido à lesão da parede do vaso sanguíneo.
Daí surgiu o modelo conhecido como Cascata, decorrente de uma sequência de fases onde cada fator leva à ativação de outro até chegar à geração da trombina, que por sua vez agindo sobre o fibrinogênio, gera fibrina.
Posteriormente foram surgindo modificações nesse Modelo da Cascata, reconhecendo-se que os fatores da coagulação circulavam na forma de pró-fatores, e somente adquiriam atividade enzimática após ativação, dependente do cálcio, e da ação de outros fatores.
Foi visto, também, que a ativação dos fatores seguia caminhos diferentes. O chamado caminho intrínseco, iniciava-se pela ativação do FXII em contato com superfícies carregadas negativamente. Daí a denominação dada, ao FXII, de fator contato. Sua ativação requer, também, a presença de outras substâncias: precalicreína (enzima) e o cininogênio de alto peso molecular (cofator sem ação enzimática).
Após a ativação do FXII, outros fatores seriam ativados, até então, seguindo essa sequência: XI, IX, VIII, X, V, II (protrombina), que atuando sobre o FI (fibrinogênio) o transforma em fibrina, que se estabiliza na presença do FXIII.
Por outro lado, uma sequência diferente dos fatores da coagulação, o caminho extrínseco da coagulação, teria início após rotura da parede vascular. Em consequência, verifica-se a exposição do fator tecidual (FT) também conhecido como (FIII) que se une ao FVII formando um complexo, o qual ativa o FX (já descrito para o caminho intrínseco), seguindo um caminho comum (também descrito anteriormente).
Esses dois caminhos unidos dão ao esquema da coagulação o aspecto da letra "Y" conforme é visto na figura 1:
Figura 1. Esquema da coagulação: HMWK = Cininogênio de alto peso molecular. PK = Pré-calicreina; TF = Fator tecidual. Maiores detalhes em relação ao modelo em cascata podem ser vistos na página desse Hemostasia (1) no menu desse site.
2 - Hemostasia primária
Vamos iniciar esse assunto apresentando, na figura 2, um desenho esquemático do início do processo hemostático (adesividade plaquetária) seguido pela agregação plaquetária.

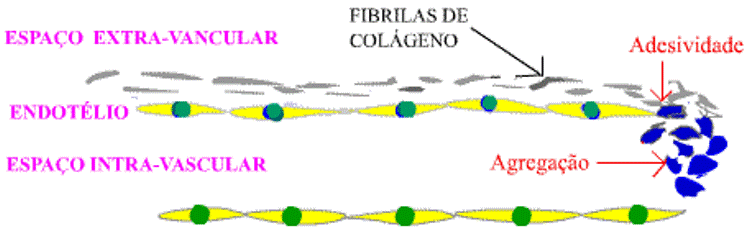
Figura 2: Início da formação do trombo hemostático.
Como é visto na figura 2, as plaquetas (em azul) aderem à fibrila de colágeno (adesividade plaquetária) e, para que a adesividade seja estável, é importante a interação com o fator "von Willebrand" (glicoproteína de adesividade).
Após a adesividade, outras plaquetas vão aderir às plaquetas unidas ao colágeno formando um aglomerado (agregação plaquetária).
Esse processo se faz em poucos segundos e é designado por hemostasia primária, reservando-se a expressão hemostasia secundária para a ativação dos fatores da coagulação que levam à formação da trombina e, consequentemente, da fibrina, pela ação da trombina sobre o fibrinogênio.
Este segundo processo demora vários minutos para ser completado e é de suma importância porque, para que o trombo hemostático seja consolidado (tornando-se efetivo para a hemostasia), ele deverá sofrer ação do ADP, colágeno e trombina.
Como dissemos anteriormente, essa subdivisão é puramente didática pois, há interdependência e simultaneidade na participação das plaquetas, dos fatores plasmáticos e da parede lesionada do vaso.
Para entendermos melhor esse texto, vamos falar, resumidamente, da estrutura da plaqueta sob o ângulo da microscopia eletrônica; assim, ficará mais fácil entendermos as explicações que se seguem. Ver Figura 3

Figura 3: A = PLAQUETA NÃO ATIVADA (Forma discoide) - 1 = Sistema canalicular aberto. 2 = Mitocôndria. 3 = Grânulos de glicogênio. 4 = Sistema tubular denso. 5 = Corpo denso. 6 = Grânulo alfa. 7 = Microfilamentos. 8 = Microtubulos circunferenciais. 9 = Membrana plaquetária. Entre a membrana plaquetária e os microtubulos circunferenciais há os filamentos submembrana. B = PLAQUETA ATIVADA (Metamorfose plaquetária) - Deformação das plaquetas e contração dos microtubulos circunferenciais e dos filamentos submembrana com fusão dos grânulos. Nota: na hemostasia primária a plaqueta se mantém na forma discoide, enquanto na hemostasia secundária ela é modificada, principalmente pela trombina, e sofre transformações (metamorfose plaquetária).
Os corpos densos contêm: cálcio, serotonina, ATP e ADP. Os grânulos alfa contêm: fator von Willebrand, fibronectina, fibrinogênio, ß-tromboglobulina, trombospondina, fator de crescimento derivado da plaqueta (PDGF), fatores da coagulação V e VIII e uma proteína neutralizadora de heparina (fator 4 plaquetário).
Além das plaquetas, a célula endotelial vascular participa intensamente para realização do trombo hemostático primário. Numa citação de Mannucci e Remuzzi, uma protease, a metaloproteinase, cliva fisiologicamente o vWF. Essa protease degrada os multímeros de tamanho "ultra-grande" de vWF, armazenados nos corpos de Weibel-Palade das células endoteliais vasculares, onde também são produzidos. [Ver artigo original]
A ação da metaloproteinase gera fragmentos de pequeno, médio e grande tamanhos que vão compor a molécula do Fator von Willebrand (vWF), que por sua vez é secretado para o sangue e para o subendotélio. No subendotélio o depósito desse fator forma a matriz que concorre para a adesividade das plaquetas.
Para complementar os dados referentes às plaquetas e ao subendotélio, vamos fazer comentários sobre os receptores que tomam parte na função plaquetária, primeiro na adesividade ao colágeno (Figuras 4 e 6) e, posteriormente, na agregação plaquetária.
Os componentes desses receptores são glicoproteínas que se associam formando complexos.

Figura 4: Participação dos receptores plaquetários e do colágeno: GP Ia/IIa=receptor da fibrila de colágeno (específico para a plaqueta). vWF=Fator von Willebrand. GP Ib/IX/V=receptor da plaqueta para o vWF. GP IIb/IIIa=receptor da plaqueta para o vWF e para o fibrinogênio. GP Ib/IX/V=receptor da plaqueta para a trombina. Detalhes no texto.
Na Figura 4 podemos ver que o complexo GP Ia/IIa funciona como receptor da fibrila de colágeno para o vWF.
Já os complexos GP Ib/IX/V e IIb/IIIa são os receptores plaquetários para o vWF. Desse modo, o vWF serve de ponte entre os citados receptores promovendo, assim, a adesividade da plaqueta ao colágeno que foi exposto após a lesão do endotélio. Nesse momento em que a plaqueta adere ao colágeno, ela é ativada e libera, além de outras substâncias, o ADP que é um ativador plaquetário.
Por outro lado, o complexo GP Ib/IX/V, também é receptor para a trombina, enzima que promoverá a ativação plaquetária juntamente com o ADP e que comentaremos no item hemostasia secundária, pois a trombina é gerada após ativação do mecanismo da coagulação.
Bonnefoy e colaboradores mostraram que o fibrinogênio também pode mediar a adesividade das plaquetas à uma superfície e entre as plaquetas. O fibrinogênio solúvel somente propicia a agregação das plaquetas quando estas estão ativadas pelos agonistas, tais como o ADP e a trombina. O receptor das plaquetas para esse fim é a GP IIb/IIIa.
Assim, o fibrinogênio faz uma ponte entre os receptores GP IIb/IIIa de duas plaquetas. Esse receptor também é usado pelo fibrinogênio adsorvido em superfície porém, essa ligação, ao contrário da anterior, não requer a ativação plaquetária.
Queremos destacar que a ligação das substâncias citadas aos respectivos receptores, e a de outras substâncias não citadas, se dá com maior ou menor facilidade na dependência de um mecanismo, que vem sendo estudado intensamente, conhecido como "shear rate/stress".
"Shear rate" corresponde à interface do sangue circulante com a parede vascular. Em outras palavras, é a velocidade de deslocamento da lâmina de sangue circulante, mais próxima à parede do vaso. As plaquetas dessa camada de sangue são, justamente, aquelas que entrarão em contato com o subendotélio exposto após a lesão do endotélio.
"Shear stress" representa a força de colisão das plaquetas entre si e da plaqueta com uma superfície, por exemplo, a fibrila de colágeno. No fenômeno de "shear rate" as plaquetas são arrastadas em uma superfície; já no "shear stress" elas se chocam com a superfície. É importante ressaltar que esse segundo fenômeno verifica-se mais intensamente quando existe formação de turbilhão. Acontece com frequência em casos patológicos, quando há formação de placas de ateroma ou quando há estrangulamento da luz do vaso.
Como mencionamos anteriormente, o "shear rate/stress" aumenta ou diminui a capacidade de uma substância ligar-se ao seu receptor. Quando diversas substâncias têm a propriedade de ligação a um mesmo receptor, a variação do grau do "shear rate/stress" facilitará ou dificultará a ligação de cada substância.
Como exemplo, citamos a interação do vWF com o complexo GP Ib/IX/V que é facilitada com a intensificação do "shear rate/stress".
Outro exemplo é a ligação entre duas plaquetas pelo fibrinogênio e o receptor GP IIb/IIIa que fica facilitada pelo "shear stress" fisiológico.

Figura 5: Esquema das reações bioquímicas que levam à geração de Tromboxane A2, de Prostaciclina e ativam a actina e a miosina, responsáveis pela contração dos microtubulos circunferenciais e dos filamentos submembrana. Detalhes no texto.
Decorrente da ação do colágeno, do ADP e da trombina, ocorrem tanto na membrana plaquetária quanto no seu interior, diversas reações que conduzem às transformações físico-químicas referidas anteriormente.
Na Figura 5 destacamos duas dessas reações: a primeira, promove a geração de tromboxane A2 pela plaqueta que leva à agregação plaquetária e de prostaciclina (PGI2), pela célula endotelial, que tem ação inversa; a segunda reação produz a metamorfose plaquetária que inclui a fusão dos grânulos, e de outros componentes, com a consequente secreção de diversas substâncias que participam do mecanismo hemostático (uma delas é o ADP).
Na primeira reação, o ácido aracdônico é liberado de dois fosfolipídios da membrana - fosfatidilinositol e fosfatidilcolina. Essa liberação depende da ação das enzimas das membranas da plaqueta e da célula endotelial, - fosfolipase C e fosfolipase A2 que são ativadas quando as substâncias, acima referidas, se ligam aos receptores correspondentes. Com a participação da enzima ciclo-oxigenase, são produzidas as endoperoxidases.
Até esse ponto as reações são comuns, tanto para as plaquetas quanto para as células endoteliais; daí em diante, nas plaquetas, forma-se o tromboxane A2 com a participação da enzima tromboxane-sintetase, e nas células endoteliais, é formada a prostaciclina com a participação da prostaciclina-sintetase.
Na segunda reação, as alterações morfológicas plaquetárias decorrem da participação da miosina e da actina, proteínas responsáveis pela contração plaquetária e a consequente secreção já referida.
Além dessas duas reações, é importante assinalar que o ADP liberado pela plaqueta promove alteração da conformação do complexo de glicoproteinas IIb/IIIa o que permite a ligação do fibrinogênio ao referido complexo e, consequentemente, a união entre plaquetas (agregação plaquetária).
Com o exposto, já temos os elementos, importantes, para desenhar o trombo hemostático primário e apresenta-lo na Figura 6.

Figura 6. Trombo hemostático primário completo. Detalhes no texto.
Como vemos na Figura 6, o trombo hemostático primário está formado pelas plaquetas aderidas à fibrila de colágeno tendo como ligante (seta 2) GP Ia/IIa do colágeno, vWF como ponte e GP Ib/IX/V da plaqueta (adesividade plaquetária). As plaquetas estão aderidas umas às outras pelo ligante (seta 1) GP IIb/IIIa das plaquetas tendo o fibrinogênio como ponte (agregação plaquetária).
Ressaltamos que esse trombo não está completo para uma hemostasia efetiva e que pode desfazer-se com facilidade. Para que ele seja completado (consolidado), são imprescindíveis as participações dos mecanismos de coagulação, anticoagulação e fibrinólise que descreveremos a seguir.
TOPO
3 - Hemostasia secundária
Considera-se como hemostasia secundária as ativações dos mecanismos de coagulação, anticoagulação e fibrinólise que vão atuar no trombo hemostático primário para torna-lo estável.
Esses três sistemas são compostos por substâncias na forma inativa e, para atuarem, necessitam ser ativadas. Para representar qualquer substância na forma ativada, usamos a letra minúscula "a". Exemplo: a forma inativa do fator X é representada por FX e a ativa por FXa.
Outra nota de valor é a de que os fatores II, VII, IX e X, ao serem fabricados no fígado, necessitam da Vit. K para torna-los aptos na ligação aos fosfolipídios com o auxílio do cálcio e, desse modo, poderem agir.
No fígado, no momento da síntese desses fatores, uma carboxilase que depende da Vit. K, catalisa uma reação de adição de um grupo carboxil em um resíduo de ácido glutâmico o que faz com que esses fatores passem a ter dois resíduos de ácido carboxiglutâmico. A importância disso é que esse duplo resíduo de ácido carboxiglutâmico liga-se ao cálcio que, por sua vez, é o responsável pela ligação do fator à superfície fosfolipídica carregada negativamente. Isso torna possível a atividade biológica do fator ativado.
É precisamente nesse ponto que os antagonistas da Vit. K (como o varfarim) atuam. Os referidos sistemas atuam concomitantemente, e permanecem em equilíbrio dinâmico nos estados de normalidade.
Como veremos, as reações de ativação se passam em grupos de poucas substâncias que denominamos por complexos. Esses complexos estão sempre associados a uma superfície - da fibrila de colágeno, da célula endotelial, da plaqueta ou do monócito.
A finalidade principal dessas reações é a de gerar trombina, que será imprescindível na ativação das plaquetas que formam o trombo primário, na geração de fibrina que reforça o trombo hemostático e na ativação dos fatores V, VIII e XIII.
É importante destacar dois setores da circulação sanguínea onde as substâncias dos mecanismos da coagulação, da anticoagulação e da fibrinólise, bem como as plaquetas, se comportam de forma diferente - o do sangue circulante, onde as substâncias e as plaquetas estão na forma inativa, e no local da injúria vascular (microambiente hemostático), onde elas são ativadas.
Mecanismo da coagulação
Na Figura 7 apresentamos um esquema global e simplificado do mecanismo da coagulação.

Figura 7. Esquema geral do mecanismo de coagulação. São destacadas cinco reações que, no texto a seguir, serão detalhadas e representadas esquematicamente.
A ativação do mecanismo de coagulação tem início por duas vias diferentes: 1 - via intrínseca e 2 - via extrínseca. Essas duas vias correspondem, respectivamente, às reações 1 e 2 descritas abaixo.
A denominação de via intrínseca deriva do fato de que na ativação tomam parte, apenas, os fatores circulantes da coagulação, enquanto na via extrínseca um fator não circulante - fator tecidual (TF), proveniente da célula endotelial, é o desencadeante.
Reação 1: conhecida por via intrínseca ou por fase de contato da coagulação. Começa quando o FXII se une à fibrila de colágeno subendotelial que fica exposta após lesão do vaso sanguíneo (Figura 8).

Figura 8. (Reação 1) HMWK=Cininogênio de alto peso molecular. PK=Pré-Calicreína. K=Calicreína
Ao se unir à fibrila de colágeno, o FXII forma um complexo com o cininogênio de alto peso molecular (HMWK) e com a pré-calicreína (PK). Na união do FXII com o HMWK e o colágeno, o FXII é ativado (FXIIa). O FXIIa ativa o FXI que, por sua vez, ativa outros fatores da coagulação. O FXIa também ativa a PK e a transforma em calicreína (K) que acelera a ativação do FXII.
É importante ressaltar que esse início da ativação da coagulação (caminho intrínseco) pelos fatores contato que levam à ativação do FXI não é o mais importante para a ativação do restante do mecanismo. A razão para isso está em que a deficiência dos fatores contato não leva a doenças hemorrágicas, e conhecemos um outro início da ativação do mecanismo da coagulação que será descrito a seguir.
Reação 2: conhecida por via extrínseca. Depende de um fator não-circulante - fator tecidual (TF) - que é uma lipoproteína que faz parte das membranas celulares.
Quando a célula endotelial sofre lesão, expõe o TF que ativa o FVII na presença do cálcio (Figura 9). Fica então formado um complexo com a participação do TF, FVII e do cálcio.

Figura 9. (Reação 2) TF=fator tecidual. Ca=cálcio.
Reação 3: Como pode ser visto na Figura 10, essa reação é o ponto de convergência das duas reações descritas anteriormente.

Figura 10. (Reação 4) Ativação da protrombina (FII). Detalhes no texto.
A geração da trombina que decorre da ação do FXa sobre a protrombina (FII), com a participação do FV e do cálcio, se faz na presença de fosfolipídio em laboratório, ou do fosfolipídio da membrana de qualquer célula. Entretanto, quando essa reação acontece na membrana plaquetária, a geração da trombina é acelerada milhares de vezes.
Reação 5: como é visto na Figura 11, a trombina é o pivô da coagulação sanguínea com ação em múltiplos pontos no processo hemostático.
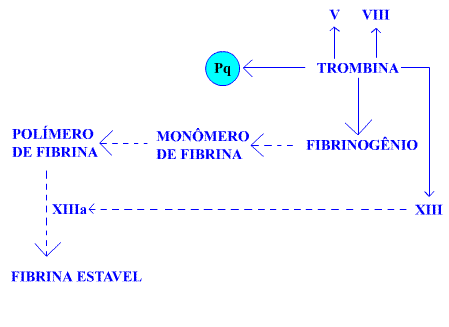
Figura 11. (Reação 5) Ação da trombina. Pq=plaqueta. Detalhes no texto.
Além da ativação das plaquetas no trombo hemostático, a trombina ativa os fatores V, VIII, XIII e atua sobre o fibrinogênio. Na ação sobre o fibrinogênio, há liberação de fibrinopeptídios A e B gerando os monômeros de fibrina. Os monômeros de fibrina ligam-se longitudinalmente e lateralmente formando os polímeros de fibrina. Essa fibrina formada é conhecida por fibrina solúvel porque, os monômeros que a formam estão ligados por pontes de hidrogênio. Com esse tipo de ligação, a fibrina dissolve-se facilmente em solução de uréia.
Nesse ponto, é importantíssima a atuação do FXIIIa pois, graças a esse fator, os monômeros de fibrina ficarão ligados covalentemente, o que confere insolubilidade à fibrina. Daí decorre a designação de fibrina estável.
Mostramos até agora como o organismo é capaz de construir um trombo hemostático efetivo. Todas as reações referidas têm lugar no microambiente hemostático e, a quantidade de trombina gerada nesse local possui o potencial para coagular todo o sangue circulante. Desse modo, a difusão da trombina, e de outros fatores ativados, precisa ser impedida para que não se estenda para a circulação sanguínea e, caso isso venha a ocorrer, é importante que essas substâncias sejam neutralizadas.
Para que tudo isso não aconteça, o organismo lança mão de importantes mecanismos que serão comentados no item sobre anticoagulação.
TOPO
4 - Anticoagulação
Ação anticoagulante da fibrina: já no próprio trombo hemostático, a fibrina apresenta intensa ação protetora impedindo que grande parte da trombina escape desse local. Para se ter idéia dessa ação protetora, a fibrina formada na coagulação de 1 ml de plasma absorve 1.000 U de trombina.
Ação protetora do fluxo sanguíneo: a trombina e os demais fatores da coagulação, que escapam do microambiente hemostático, são diluídos na corrente sanguínea o que os impede de agir e, além disso, os tornam vulneráveis à inativação.
Ação protetora do sistema reticuloendotelial: as células do sistema reticuloendotelial afastam da circulação, por meio da fagocitose, qualquer material com capacidade de ativar os fatores da coagulação (material tromboplástico), procedendo do mesmo modo com a fibrina e com seus produtos de degradação.
Inibidores da coagulação (anticoagulantes): os principais são a Antitrombina III, as Proteínas C e S e o Inibidor da Via do Fator Tecidual (TFPI). Ver Figura 12.

Figura 12. Em azul os fatores da coagulação e em vermelho os anticoagulantes. SH=Sulfato de heparam. TFPI=Inibidor da via do fator tecidual. TF=Fator tecidual. ATIII=Antitrombina III. TM=Trombomodulina. PC=Proteína C. PS=Proteína S.
A ATIII ao formar complexos com vários fatores da coagulação, particularmente com os fatores II, IX, X e XI, os inativa. Essa inativação é potencializada pela heparina e pelo sulfato de heparam (molécula semelhante à heparina que está presente na superfície da célula endotelial).A PC torna-se uma protease ativa ao formar um complexo com a TM (proteína presente na superfície da célula endotelial) e o FIIa (trombina), sendo auxiliada pelo cofator PS. A PCa promove a proteólise dos fatores V e VIII inibindo, desse modo, a geração de trombina. O TFPI forma um complexo com o TF e o FVII que inibe o FX.
5 - Fibrinólise
Por fim, após a ação protetora dos anticoagulantes, a fibrina que for gerada fora do microambiente hemostático e a fibrina do trombo hemostático, após cumprida sua função, serão lisadas pela ativação do mecanismo fibrinolítico. As enzimas, com função ativadora, que tomam parte no mecanismo fibrinolítico, são serina-proteases por apresentarem um sítio ativo composto pelos aminoácidos: serina, ácido aspártico e histidina. Já os inibidores do sistema fibrinolítico, são membros da superfamília "serpin" (serine proteinase inhibitor). Elas têm como característica um sítio reativo específico (Arg-X ou Lys-X) que, após clivagem pela sua enzima alvo, leva à formação de um complexo formado pelo inibidor acoplado à enzima alvo. O agente principal do sistema fibrinolítico é a plasmina. Segundo Collen, a designação de sistema fibrinolítico é inadequada; isso porque a fibrinólise é apenas uma resultante da atividade da plasmina. Outra resultante, também de grande importância, decorre de sua ação sobre as matrizes metaloproteinases (MMPs) que têm a propriedade de degradar a matriz extracelular (ECM).Collen sugere a designação de Sistema do Plasminogênio ao invés de Sistema Fibrinolítico.
Como pode ser visto na Figura 13, a plasmina é gerada a partir do plasminogênio após ação dos ativadores do plasminogênio: ativador do plasminogênio de tipo tecidual (t-PA) e ativador do plasminogênio de tipo uroquinase (u-PA). Na geração da plasmina pelo u-PA, esse ativador se liga ao seu receptor celular u-PAR que leva à formação da MMP responsável pelos fenômenos de migração celular e remodelação tecidual.

Figura 13. Em verde representamos os componentes de ativação e em vermelho os de inibição. t-PA=Ativador do plasminogênio de tipo tecidual. u-PA=Ativador do plasminogênio de tipo uroquinase. u-PAR=Receptor celular do u-PA. Alfa2-AP=Alfa2-Antiplasmina. FDP=Produto de degradação da fibrina. PAI=Inibidor dos ativadores do plasminogênio. MMP=Matriz metaloproteinase. ECM=Matriz extracelular. TIMP=Inibidor tecidual da MMP.
Entretanto, a reação que se relaciona ao mecanismo hemostático, e que vamos descrever, é a da fibrinólise. Nessa reação o plasminogênio é transformado em plasmina pela ação do t-PA. A plasmina, ao atuar sobre a fibrina, gera os produtos de degradação da fibrina (FDP).
Há distinção na intensidade de ação dos componentes do sistema fibrinolítico na dependência do meio em que atuam, se na superfície da fibrina (fase sólida) ou no sangue circulante (fase líquida). Na Figura 14 representamos na parte superior, a fase líquida e na inferior, a fase sólida.

Figura 14. PG=Plasminogênio. FDP=Produto de degradação da fibrina. t-PA=Ativador, de tipo tecidual, do plasminogênio. Alfa2-AP=Alfa2-Antiplasmina.
O t-PA apresenta alta afinidade pelo plasminogênio na superfície da fibrina e baixa afinidade no sangue circulante. Isso parece decorrer da facilitação da ligação do t-PA ao plasminogênio pela fibrina.
O t-PA e o seu inibidor (PAI) são sintetizados e secretados pelas células endoteliais e são depurados, rapidamente, pelo fígado. A célula endotelial tem a propriedade de promover a ligação do plasminogênio e do t-PA à sua superfície e, por via do Anexin II, impede a reação das duas substâncias.
O plasminogênio também se liga à membrana de outros tipos de células, além da célula endotelial, entretanto, a lipoproteina(a) [Lp(a)] compete com o plasminogênio nessa ligação, o que impede sua ativação na superfície celular.
O t-PA ativa o plasminogênio na superfície da fibrina com intensidade 100 vezes maior do que no sangue circulante (fase líquida), o que colabora para confinar a ação da plasmina, em casos de normalidade, onde a fibrina é formada.
Por outro lado, a plasmina gerada e fixada na superfície da fibrina é protegida da rápida inativação por parte da Alfa2-AP, o que não se verifica na fase líquida.
Há muito tempo é sabido que a trombina possui ação inibitória sobre a fibrinólise.
Essa ação inibitória é exercida por uma enzima que decorre da ativação de um zimogênio (proenzima) pela trombina, tendo como cofator a trombomodulina.
Essa enzima foi identificada, recentemente, por cinco grupos de pesquisadores, independentemente. Dentre esses pesquisadores está Laszlo Bajzar que se refere à molécula, com tal propriedade, como "thrombin activatable fibrinolysis inhibitor (TAFI). Em alguns artigos a palavra "activatable" é substituída por "activable".
A ativação do TAFI acontece após a formação de um complexo ternário composto pela trombina (enzima)/trombomodulina (cofator) / TAFI (substrato).
É sabido que o plasminogênio se liga às lisinas do carbono terminal da fibrina e, desse modo, adquire uma conformação que possibilita sua maior ativação.